Code
Rendered code
Output files
I’ve already used miRanda to predict miRNA-lncRNA binding. Now I want to assess coexpression for miRNA-lncRNA pairs using Pearson’s correlation coefficient.
#library(energy)
library(tidyr)
library(dplyr)
library(readr)
library(ggplot2)Now that we’ve found putative interactions, including those with high complementarity, in 10-Apul-lncRNA-miRNA-miRanda we need to validate by examining patterns of coexpression. We’d expect a putatively-interacting miRNA-lncRNA pair to be highly coexpressed, and we’d expect a strong negative relationship to indicate either a lncRNA sponging miRNA OR an miRNA targeting lncRNA for degradation.
0.1 Obtain Pearson’s coefficient correlation values
Read in, format, and normalize data
miRNA_counts <- read.delim("../output/03.10-D-Apul-sRNAseq-expression-DESeq2/Apul_miRNA_ShortStack_counts_formatted.txt")
# Simplify column names
colnames(miRNA_counts) <- sub("_.*", "", colnames(miRNA_counts))
colnames(miRNA_counts) <- sub("X", "", colnames(miRNA_counts))
# Remove any miRNA with 0 counts across samples
miRNA_counts<-miRNA_counts %>%
mutate(Total = rowSums(.[, 1:5]))%>%
filter(!Total==0)%>%
dplyr::select(!Total)
lncRNA_counts_full <- read.delim("../output/08-Apul-lncRNA/counts.txt", skip=1)
rownames(lncRNA_counts_full) <- lncRNA_counts_full$Geneid
lncRNA_counts <- lncRNA_counts_full %>% select(-Geneid, -Chr, -Start, -End, -Strand, -Length)
# Format lncRNA column names to match the miRNA names
colnames(lncRNA_counts) <- sub("...data.", "", colnames(lncRNA_counts))
colnames(lncRNA_counts) <- sub(".sorted.bam", "", colnames(lncRNA_counts))
# Order lncRNA column names to match the miRNA column order
lncRNA_counts <- lncRNA_counts[, colnames(miRNA_counts)]
# Check that the columns match name and order for both dataframes
colnames(lncRNA_counts) == colnames(miRNA_counts)## [1] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
## [16] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
## [31] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE# Remove any lncRNAs with 0 for all samples
lncRNA_counts <- lncRNA_counts %>%
mutate(Total = rowSums(.[, 1:5]))%>%
filter(!Total==0)%>%
dplyr::select(!Total)
# Function to normalize counts (simple RPM normalization)
normalize_counts <- function(counts) {
rpm <- t(t(counts) / colSums(counts)) * 1e6
return(rpm)
}
miRNA_norm <- normalize_counts(miRNA_counts)
lncRNA_norm <- normalize_counts(lncRNA_counts)# Function to calculate PCC and p-value for a pair of vectors
calc_pcc <- function(x, y) {
result <- cor.test(x, y, method = "pearson")
return(c(PCC = result$estimate, p_value = result$p.value))
}
# Create a data frame of all miRNA-lncRNA pairs
pairs <- expand.grid(miRNA = rownames(miRNA_norm), lncRNA = rownames(lncRNA_norm))
# Calculate PCC and p-value for each pair
pcc_results <- pairs %>%
rowwise() %>%
mutate(
pcc_stats = list(calc_pcc(miRNA_norm[miRNA,], lncRNA_norm[lncRNA,]))
) %>%
unnest_wider(pcc_stats)
# Adjust p-values for FDR
pcc_results <- pcc_results %>%
mutate(adjusted_p_value = p.adjust(p_value, method = "fdr"))
# Save as csv
write.csv(pcc_results, "../output/14.2-Apul-miRNA-lncRNA-coexpression/Apul-PCC_miRNA_lncRNA-full.csv")Check
# Read in results
pcc_results <- read.csv("../output/14.2-Apul-miRNA-lncRNA-coexpression/Apul-PCC_miRNA_lncRNA-full.csv")
# Use this code to download the PCC results if needed
#pcc_results <- read.csv("https://gannet.fish.washington.edu/kdurkin1/ravenbackups/timeseries_molecular/D-Apul/output/14.2-Apul-miRNA-lncRNA-coexpression/Apul-PCC_miRNA_lncRNA-full.csv")
nrow(pcc_results)## [1] 950283nrow(pcc_results%>% filter(abs(PCC.cor) > 0.90))## [1] 3nrow(pcc_results %>% filter(p_value < 0.05))## [1] 81605nrow(pcc_results %>% filter(p_value < 0.05 & abs(PCC.cor) > 0.90))## [1] 3of the 950,283 possible miRNA-lncRNA interactions, 3 have a Pearson’s correlation coefficient with a magnitude above 0.9, and 81,605 have a significant correlation (pval<0.05). All of the coefficients with a magnitude >0.9 are significant.
I find it interesting that so many putative interactions have significant pvalues, but so few have correlation coefficients above 0.9. What does the distribution of significant correlation coefficients look like?
ggplot(pcc_results[pcc_results$p_value < 0.05,], aes(x = PCC.cor)) +
geom_density(fill = "blue", alpha = 0.5) +
labs(title = "Density of Significant Correlations",
x = "Correlation Coefficient",
y = "Density") +
theme_minimal()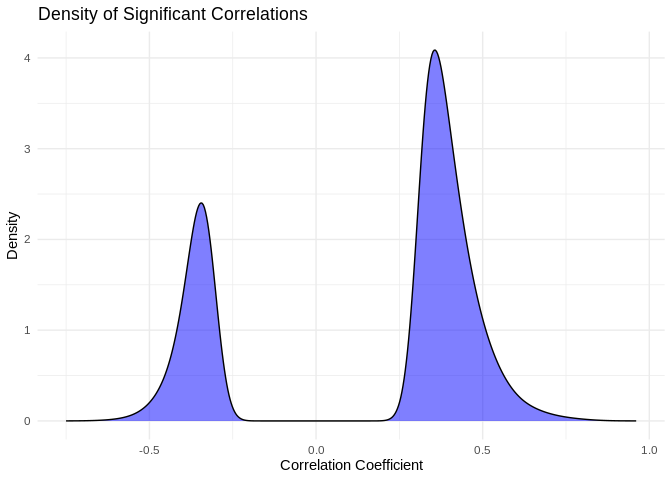
ggplot(pcc_results[pcc_results$p_value < 0.05,], aes(x = abs(PCC.cor))) +
geom_density(fill = "blue", alpha = 0.5) +
labs(title = "Density of Significant Correlations (magnitudes)",
x = "Magnitude of Correlation Coefficient",
y = "Density") +
theme_minimal()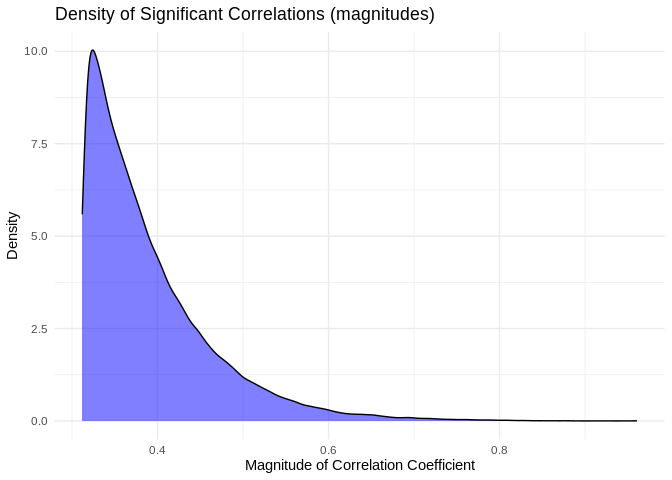
Mean of the absolute value of correlation for significant correlation coefficients is {r} round(mean(abs(pcc_results[pcc_results$p_value < 0.05,]$PCC.cor)),2)
1 Incorporate miRanda target prediction results
1.1 full lncRNA
I realized that the lncRNA counts matrix includes some duplicate lncRNA, where two or more distinct lncRNA IDs share the exact same genomic coordinates. As far as I can tell all of the duplicate lncRNAs have counts of 0 across all samples, so it was likely just a naming error. This also means the duplicates didn’t affect the correlation calculations above, since I filtered out lncRNAs that had all-0 counts before PCC.
I’ll need to filter then out though to facilitate joining.
# Create mapping table that shows the full genomic location for each lncRNA
lncRNA_mapping <- data.frame(
lncRNA = lncRNA_counts_full$Geneid,
location = paste0(lncRNA_counts_full$Chr, ":", (lncRNA_counts_full$Start-1), "-", lncRNA_counts_full$End)
)
# Isolate lncRNA that have been repeated
repeats <- lncRNA_mapping[duplicated(lncRNA_mapping$location) | duplicated(lncRNA_mapping$location, fromLast = TRUE), ]
# Isolate the repeated lncRNA counts
repeats_counts <- lncRNA_counts_full %>% filter(Geneid %in% repeats$lncRNA)
nrow(lncRNA_mapping)## [1] 24181length(unique(lncRNA_mapping$location))## [1] 23782nrow(lncRNA_mapping) - length(unique(lncRNA_mapping$location))## [1] 399head(repeats_counts)## Geneid Chr Start End Strand Length
## lncRNA_062 lncRNA_062 ntLink_6 7465722 7467150 + 1429
## lncRNA_063 lncRNA_063 ntLink_6 7465722 7467150 + 1429
## lncRNA_071 lncRNA_071 ntLink_6 7548691 7551215 + 2525
## lncRNA_072 lncRNA_072 ntLink_6 7548691 7551215 + 2525
## lncRNA_080 lncRNA_080 ntLink_6 9559991 9569239 + 9249
## lncRNA_081 lncRNA_081 ntLink_6 9559991 9569239 + 9249
## ...data.1A10.sorted.bam ...data.1A12.sorted.bam
## lncRNA_062 0 0
## lncRNA_063 0 0
## lncRNA_071 0 0
## lncRNA_072 0 0
## lncRNA_080 0 0
## lncRNA_081 0 0
## ...data.1A1.sorted.bam ...data.1A2.sorted.bam ...data.1A8.sorted.bam
## lncRNA_062 0 0 0
## lncRNA_063 0 0 0
## lncRNA_071 0 0 0
## lncRNA_072 0 0 0
## lncRNA_080 0 0 0
## lncRNA_081 0 0 0
## ...data.1A9.sorted.bam ...data.1B10.sorted.bam
## lncRNA_062 0 0
## lncRNA_063 0 0
## lncRNA_071 0 0
## lncRNA_072 0 0
## lncRNA_080 0 0
## lncRNA_081 0 0
## ...data.1B1.sorted.bam ...data.1B2.sorted.bam ...data.1B5.sorted.bam
## lncRNA_062 0 0 0
## lncRNA_063 0 0 0
## lncRNA_071 0 0 0
## lncRNA_072 0 0 0
## lncRNA_080 0 0 0
## lncRNA_081 0 0 0
## ...data.1B9.sorted.bam ...data.1C10.sorted.bam
## lncRNA_062 0 0
## lncRNA_063 0 0
## lncRNA_071 0 0
## lncRNA_072 0 0
## lncRNA_080 0 0
## lncRNA_081 0 0
## ...data.1C4.sorted.bam ...data.1D10.sorted.bam
## lncRNA_062 0 0
## lncRNA_063 0 0
## lncRNA_071 0 0
## lncRNA_072 0 0
## lncRNA_080 0 0
## lncRNA_081 0 0
## ...data.1D3.sorted.bam ...data.1D4.sorted.bam ...data.1D6.sorted.bam
## lncRNA_062 0 0 0
## lncRNA_063 0 0 0
## lncRNA_071 0 0 0
## lncRNA_072 0 0 0
## lncRNA_080 0 0 0
## lncRNA_081 0 0 0
## ...data.1D8.sorted.bam ...data.1D9.sorted.bam ...data.1E1.sorted.bam
## lncRNA_062 0 0 0
## lncRNA_063 0 0 0
## lncRNA_071 0 0 0
## lncRNA_072 0 0 0
## lncRNA_080 0 0 0
## lncRNA_081 0 0 0
## ...data.1E3.sorted.bam ...data.1E5.sorted.bam ...data.1E9.sorted.bam
## lncRNA_062 0 0 0
## lncRNA_063 0 0 0
## lncRNA_071 0 0 0
## lncRNA_072 0 0 0
## lncRNA_080 0 0 0
## lncRNA_081 0 0 0
## ...data.1F11.sorted.bam ...data.1F4.sorted.bam
## lncRNA_062 0 0
## lncRNA_063 0 0
## lncRNA_071 0 0
## lncRNA_072 0 0
## lncRNA_080 0 0
## lncRNA_081 0 0
## ...data.1F8.sorted.bam ...data.1G5.sorted.bam
## lncRNA_062 0 0
## lncRNA_063 0 0
## lncRNA_071 0 0
## lncRNA_072 0 0
## lncRNA_080 0 0
## lncRNA_081 0 0
## ...data.1H11.sorted.bam ...data.1H12.sorted.bam
## lncRNA_062 0 0
## lncRNA_063 0 0
## lncRNA_071 0 0
## lncRNA_072 0 0
## lncRNA_080 0 0
## lncRNA_081 0 0
## ...data.1H6.sorted.bam ...data.1H7.sorted.bam ...data.1H8.sorted.bam
## lncRNA_062 0 0 0
## lncRNA_063 0 0 0
## lncRNA_071 0 0 0
## lncRNA_072 0 0 0
## lncRNA_080 0 0 0
## lncRNA_081 0 0 0
## ...data.2B2.sorted.bam ...data.2B3.sorted.bam ...data.2C1.sorted.bam
## lncRNA_062 0 0 0
## lncRNA_063 0 0 0
## lncRNA_071 0 0 0
## lncRNA_072 0 0 0
## lncRNA_080 0 0 0
## lncRNA_081 0 0 0
## ...data.2C2.sorted.bam ...data.2D2.sorted.bam ...data.2E2.sorted.bam
## lncRNA_062 0 0 0
## lncRNA_063 0 0 0
## lncRNA_071 0 0 0
## lncRNA_072 0 0 0
## lncRNA_080 0 0 0
## lncRNA_081 0 0 0
## ...data.2F1.sorted.bam ...data.2G1.sorted.bam
## lncRNA_062 0 0
## lncRNA_063 0 0
## lncRNA_071 0 0
## lncRNA_072 0 0
## lncRNA_080 0 0
## lncRNA_081 0 0There are 399 duplicate lncRNAs in the counts data
Since I’ll be using lncRNA_mapping for joining purposes, it must contain only unique genomic locations.
Deduplicate lncRNA_mapping:
unique_mapping <- lncRNA_mapping[!duplicated(lncRNA_mapping$location), ]
nrow(lncRNA_mapping) - nrow(unique_mapping)## [1] 399Great! Now that we have a mapping file of only our unique lncRNAs, we can proceed to incorporating the miRanda output.
# miRNA-lncRNA_full miRanda output
miRNA_lncRNA_miRanda <- read_delim("../output/10-Apul-lncRNA-miRNA-miRanda/Apul-miRanda-lncRNA-strict-parsed.txt", col_names=FALSE)## Rows: 178148 Columns: 9
## ── Column specification ────────────────────────────────────────────────────────
## Delimiter: "\t"
## chr (6): X1, X2, X5, X6, X8, X9
## dbl (3): X3, X4, X7
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.colnames(miRNA_lncRNA_miRanda) <- c("mirna", "Target", "Score", "Energy_Kcal_Mol", "Query_Aln", "Subject_Aln", "Al_Len", "Subject_Identity", "Query_Identity")
# format miRNA names
miRNA_lncRNA_miRanda$mirna <- gsub(">", "", miRNA_lncRNA_miRanda$mirna)
miRNA_lncRNA_miRanda$mirna <- gsub("\\..*", "", miRNA_lncRNA_miRanda$mirna)
# Join with mapping file to annote miRanda results with lncRNA IDs
miRNA_lncRNA_miRanda <- left_join(miRNA_lncRNA_miRanda, unique_mapping, by=c("Target" = "location"))
# Create a columns that contains both the miRNA and interacting lncRNA
pcc_results$interaction <- paste0(pcc_results$miRNA, ":", pcc_results$lncRNA)
miRNA_lncRNA_miRanda$interaction <- paste0(miRNA_lncRNA_miRanda$mirna, ":", miRNA_lncRNA_miRanda$lncRNA)
# Annotate miRanda results w PCC info
miRNA_lncRNA_miRanda <- left_join(miRNA_lncRNA_miRanda, pcc_results, by="interaction")# Filter to high complementarity putative targets
target_21bp <- miRNA_lncRNA_miRanda[miRNA_lncRNA_miRanda$Al_Len > 20,]
target_21bp_3mis <- target_21bp[target_21bp$Subject_Identity>85,]
# How many w significant correlation?
nrow(miRNA_lncRNA_miRanda)## [1] 178148nrow(miRNA_lncRNA_miRanda %>% filter(!is.na(PCC.cor)))## [1] 115512nrow(miRNA_lncRNA_miRanda %>% filter(p_value < 0.05))## [1] 9677nrow(target_21bp %>% filter(p_value < 0.05))## [1] 1396nrow(target_21bp_3mis %>% filter(p_value < 0.05))## [1] 0For miRNA binding to lncRNA, miRanda predicts 178,148 putative interactions. Of these, 115,512 yield a correlation value (indicating the lncRNA is actually present in our counts data); 9,677 have significant PCCs; 1,396 are >21bp and have signficant PCCs; and 0 are >21bp with <=3 mismatches and have significant PCCs.
2 Summary
How does different input and/or complementarity filtering affect # putative interactions:
Reminder summary of miRanda results:
| Input | All | 21bp | 21bp, >=3 mismatch |
|---|---|---|---|
| lncRNA | 178,148 | 24,018 (13.48% of total) | 22 (0.012% of total) |
For different filters, how many putative interactions also show significant coexpression (PCC pval < 0.05)?
| Input | All | 21bp | 21bp, >=3 mismatch |
|---|---|---|---|
| lncRNA | 105,835 | 14,503 (13.7% of total) | 13 (0.012% of total) |
Note that some putative interactions indicated by miRanda are not present in the counts data (i.e. the miRNA and/or lncRNA had 0 counts inour RNAseq data), and are thus excluded from the PCC-filtered data
Is there a clear “cutoff” for what complementarity parameters are most associated with significant coexpression?
significant_df <- miRNA_lncRNA_miRanda %>% filter(p_value < 0.05)
# Plot with jitter (significant coexpression only)
ggplot(significant_df, aes(x = Al_Len,
y = as.numeric(gsub("%", "", Subject_Identity)))) +
geom_density_2d_filled(contour_var = "density", alpha = 0.8) +
scale_x_continuous(breaks = seq(min(significant_df$Al_Len), max(significant_df$Al_Len), by = 1)) +
labs(x = "Alignment Length (Al_Len)",
y = "Subject Identity (%)",
title = "2D Density: Alignment Length vs. Subject Identity (p < 0.05)") +
theme_minimal()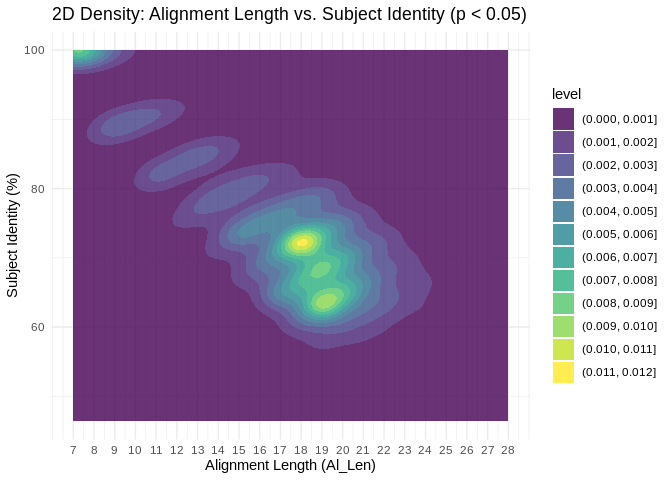 Interesting! There are significant coexpressions happening across both metrics of complementarity, but we see clustering between 17 and 20nt alignment length.
Interesting! There are significant coexpressions happening across both metrics of complementarity, but we see clustering between 17 and 20nt alignment length.
In other words, for all the miRNA-lncRNA binding interactions predicted by miRanda, interactions with high alignment lengths (17-20nt) appear most likely to also show significantly correlated expression.
There’s also a cluster at the top left corner, showing us that many interactions significant correlations have low alignment lengths but perfect complementarity. this likely represents perfect binding in the seed region.
# Save putative interactions with significantly correlated coexpression for visualization
# (e.g. creating an interaction network plot in Cytoscape)
write.csv(significant_df, "../output/14.2-Apul-miRNA-lncRNA-coexpression/miRanda-PCC-significant-miRNA_lncRNA.csv", row.names = FALSE)